Абстракт
Прогрессирование заболевания почек скоррелировано со степенью почечного внутритканевого фиброза. Проксимальные трубчатые клетки (ПТК) способствуют этим патологическим изменениям производством цитокинов и изменениями в составе внеклеточного матрикса. Гиалуронан (ГК) - присутствующий повсеместно в соединительной ткани полисахарид, который регулирует функцию клетки и поэтому может способствовать и регулировать реконструкцию ткани. В контексте диабетической нефропатии, увеличенное производство почечного ГК вовлечено в клубочковую гиперячеистость в стрептозотоциновой модели диабета. Недавние исследования также включали в себя проникновение макрофага в патогенез диабетической нефропатии. В текущем исследовании мы изучали производство ПТК ГК в ответ на изменения в концентрации глюкозы и произведенного макрофагом цитокина IL-1?.
Результаты демонстрируют временную зависимость увеличения концентрации ГК в культуре супернатанта и в ответ на 25 ммоль D-глюкозы, и в ответ на IL-1?. Экспрессия мРНК гиалуронан-синтаз (ГКС) была исследована при помощи метода ПЦР в реальном времени. ГКС2 мРНК индукция была замечена или после добавления 25 ммоль D-глюкозы или после возбуждения IL-1?. ГКС3 мРНК в обязательном порядке экспрессировалась ПТК, но не находилась в зависимости от добавления или 25 ммоль D-глюкозы или IL-1?. В контрасте экспрессия ГКС1 мРНК не была обнаружен ни в нестимулируемых, ни в стимулируемых клетках. Стимулирование производства ГК или IL-1?, или 25 ммоль D-глюкозы было аннулировано ингибированием I-каппа киназы В (ИККБ), используя Sulindac, и при помощи протеосомного ингибитора PSI, таким образом, вовлекая активацию NF-?В в механизм транскрипционной активации ГКС2.
Введение
Цель нашей работы состоит в том, чтобы идентифицировать факторы, которые в комбинации с гипергликемией, могут внести вклад и в инициирование, и в прогрессию диабетической почечной болезни. Теперь очевидно, что прогрессирующее снижение почечной функции при диабете довольно близко коррелировано со степенью почечного промежуточного фиброза. Поэтому мы сосредоточились на тех механизмах, которые могут вызвать внутритканевые изменения, и в особенности механизмы, которыми проксимальные трубчатые эпителиальные клетки (ПТК) могут быть вовлечены в их инициирование.
Недавние исследования включали в себя проникновение макрофага в патогенез диабетической нефропaтии. Поэтому, в текущем исследовании мы рассматривали регулирование ПТК ГК-генерации, в ответ на получение макрофагом цитокинов и поднятие концентрации глюкозы.
Материалы и методы
Клеточная культура: Эксперименты были выполнены, используя линию клеток HK2, (человеческие почечные проксимальные трубчатые эпителиальные ячейки, увековеченные преобразованием с человеческим вирусом папилломы [HPV] 16 генов E6/E7) при стандартных условиях культивирования. Клетки были выращены до прекращения слияния и роста в свободной сывороточной среде в течение 48 часов, и все эксперименты выполнялись в условиях без сыворотки. Клетки, прекратившие рост, стимулировались рекомбинантным цитокином IL-1? (от 0 до 100 нг/мл) или 25 ммоль D-глюкозы. Образцы надосадка были собраны вплоть 96-тичасовой длительности стимуляции для определения количества ГК. Экспериментальные образцы для контроля были выполнены добавлением 25 ммоль L-глюкозы, или 5-ммоль D-глюкозы.
Изменение в синтезе ГК: Во всех экспериментах концентрация ГК в надосадке клеточной культуры была определена связанным путём иммуносорбентации фермента (ГК «Chugai» количественный испытательный комплект, Chugai Diagnostics). Эффект различных стимулов на экспрессию ГКС1, 2 и 3 мРНК экспрессия была определена обратной транскрипцией и ПЦР (ПЦР в реальном времени) как было ранее описано, используя определенные олигонуклеотидные праймеры. Чтобы подтвердить, что производство ГК зависит от индукции транскрипции ГКС, клетки линии HK-2 стимулировались IL-1? (1 нг/мл) или 25 ммоль D-глюкозой, в присутствии возрастающих (нетоксичных) доз или актиномицина-D (0-250 нг/млl), чтобы ингибировать транскрипцию, или циклогексимида (0-5 мкг/мл), чтобы ингибировать мРНК трансляцию соответственно.
Вовлечение NF-?В: Зависимость производства ГК от активации NF-каппа В была определена ингибированием I-каппы киназы В (ИККБ), используя Sulindac, и при помощи протеосомного ингибитора PSI (Calbiochem)
Результаты
Изменения в производстве ГК: Добавление или IL-1?, или 25-ммоль D-глюкозы привело к увеличению временной зависимости производства ГК (рисунок 1). Эффект IL-1? был существенным на точке в 12 часов. По контрасту эффект 25-ммоль D-глюкозы был замедлен и стал очевидным только спустя 96 часов после ее добавления (рисунок 1). Увеличение синтеза ГК после добавления 25-ммоль D-глюкозы не было связано с изменениями в осмотическом давлении, так как добавление 25-ммоль L-глюкозы не затрагивало производство ГК. Совместная стимуляция клеток IL-1? и 25-ммоль D-глюкозы привело к аддитивному эффекту, а не к прогрессирующему увеличению производства ГК.
Индукция ГКС2 мРНК была замечена после стимуляции IL-1? (Рис. 2a). ГКС3 мРНК неизменно экспрессировалась в клетках HK-2, и не подвергалась изменениям при добавлении IL-1?. В контрасте с этим, экспрессия ГКСl мРНК не была обнаружена как в нестимулируемых, так в стимулируемых клетках. Ингибирование de novo генной транскрипции актиномицина-D или трансляции мРНК добавлением циклогексимида полностью аннулировало производство ГК после добавления IL-1?.
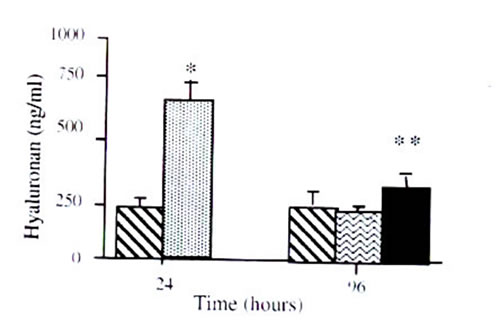
Иллюстрация 1: Стимуляция ГК в HK-2 клетках. В качестве стимула IL-1? (1 нг/мл), 25 M D-глюкоза, 25 ммоль L глюкоза, или 5-ммоль D-глюкоза были добавлены к сливающимся HK-2 клеткам.

Иллюстрация 2: Экспрессия ГК-синтазы - после добавления IL-1? [A] (l нг/мл) к сливающимся монослоям HK-2 клеток, общее количество мРНК было извлечено и экспрессия ГКС2 мРНКбыла исследована методом ПЦР в реальном времени в пунктах времени до 24 часов. В качестве контрольного эксперимента была добавлена 5-ммоль D-глюкоза (c) в отсутствии IL-1?. Похожее добавление 25-ммоль D-глюкозы [B] РНК было произведено для проведения ПЦР в реальном времени в пунктах времени до 72 часов, в этих экспериментах 5 ммоль D-глюкозы (c), или 25 ммоль L-глюкозы (25L) служили как контроль. Продукты ПЦР были отделены электрофорезом на 3%-ом агарозном геле. Один представительный эксперимент показывает для каждого из этих двух стимулов.
Добавление 25 ммоль D-глюкозы оказывало подобные эффекты на экспрессию ГК-синтазы, с индукцией экспрессии ГКС2 мРНК, но никакого эффекта на основополагающую экспрессию ГКС3 мРНК (рис. 2b). Так же добавление 25 ммоль D-глюкозы не вызывало экспрессии ГКС1 мРНК, которая снова оставалась необнаружимой методом ПЦР в реальном времени. Эффект глюкозы на производство ГК, был также ингибирован добавлением или актиномицина-D или циклогексимида.
Механизм ГК стимуляции: И IL-1?, и 25 ммоль D-глюкозы вызывали изменения в производстве ГК в клетках HK-2, которые были аннулированы добавлением Sulindac в дозозависимой манере (Рис. 3). В контрасте, индометацин не имел никакого эффекта на производство ГК после добавления любого из стимулов. Ингибирование NF-каппа активации В добавлением протеосомного ингибитора PSI также аннулировало и IL-1?, и 25 ммоль D-глюкозное стимулирование увеличения производства ГК (Рис. 4).
Обсуждение
Данные, представленные в текущей рукописи, демонстрируют, что производство ГК проксимальными трубчатыми эпителиальными клетками стимулировалось и 25 ммоль D-глюкозы, и провоспалительным цитокином IL-1?. Кроме того, полученные данные демонстрируют, что стимуляция синтеза ГК зависит от транскрипционной активации ГКС2. Различная "кинетика" стимуляции ГК после дополнения любого стимула, однако предполагает, что механизмы, которыми они добиваются этих эффектов, могут отличаться. В дополнение к индуцибeльной экспрессии ГКС2 мРНК мы также продемонстрировали обязательную экспрессию ГКС3 мРНК, который не был под влиянием дополнения или IL-1? или 25 ммоль D-глюкозы. Предыдущие исследования отмечали изменения в синтезе ГК при патогенезе клубочковых отклонений, связанных с диабетической нефропaтией. В этих исследованиях изменения в производстве ГК были связаны с изменением круговорота простагландина.
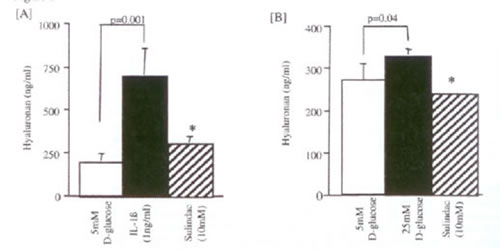
Иллюстрация 3: Sulindac ингибирует IL-1? [A] и 25 ммоль D-глюкоза [B] стимулируемое производство ГК. Сливающиеся монослои HK-2 клеток стимулировались или с l нг/мл IL-1? или с 25 ммоль D-глюкозы в присутствии sulindac. Образцы надосадка были собраны через 24 часа после добавления IL-1? и через 96 часов после следующего добавления 25 ммоль D-глюкозы для определения концентрации ГК. Данные представляют значения ± sd от 4 индивидуальных экспериментов, *p <0.005 ингибитора против IL-1? [A] или 25 ммоль D-глюкозы [B].
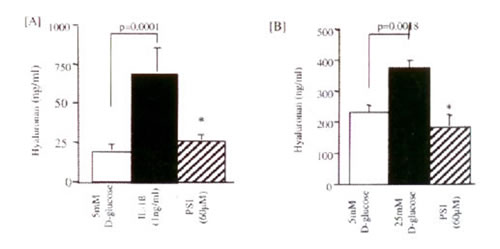
Иллюстрация 4: Ингибирование производства ГК в ответ на добавление или IL-1? [A], или 25 ммоль D-глюкозы протеосомным ингибитором PSI. Сливающиеся монослои HK-2 клеток стимулировались или с IL-1? [A], или с 25 ммоль D-глюкозы в присутствии PSI. Эксперименты для контроля были выполнены стимуляцией с 5 ммоль D-глюкозы в отсутствии PSI. Образцы супернатанта были собраны через 24 часа после дополнения IL-1? и через 96 часов после следующего добавления 25 ммоль D-глюкозы для определения концентрации ГК. Данные представляют значения ± sd от 4 индивидуальных экспериментов, *p <0.005 для PSI или против IL-1? [A], или против 25-миллиметровой D-глюкозы [B].
Хотя эффекты и IL-1?, и 25 ммоль D-глюкозы ингибируются Sulindac, маловероятно, что это связано с изменением в круговороте простагландина, поскольку ни IL-1?, ни 25 ммоль D-глюкозы вызванные изменения в синтезе ГК не были ингибированы метиндолом. Недавние исследования предполагают, что Sulindac ингибирует активацию NF-каппы В запрещением деятельности киназы IKKB. Наши результаты демонстрируют, что индукция ГК или IL-1?, или 25 ммоль D-глюкозы была связана с активацией NF-каппы B. Кроме того, ингибирование активации NF-каппы В или с помощью Sulindac, или при помощи протеосомного ингибитора прекращало производство ГК в ответ на любой стимул. Путь NF-каппа В регулирует клеточный ответ на множество стимулов, включая цитокины, бактериальную и вирусную инфекцию и активацию клеточных путей стресса. Это также важно для контроля клеточного роста и, как было продемонстрировано, является важным регулятором апоптоза. Его активация, как известно, вызвана многочисленными провоспалительными цитокинами, такими как IL-1?. Это - первая демонстрация того, что активация NF-каппы В, увеличивает синтез ГК транскрипционной активацией ГКС2, хотя транскрипционная активация ГКС1 TNFa, была недавно продемонстрирована в линии клеток миофибробластов. Активация NF-каппы В повышенной концентрацией глюкозы была недавно продемонстрирована в сосудистых гладких мышечных клетках. Также наши данные являются первыми, которые демонстрируют активацию NF-каппы В после добавления 25 ммоль D-глюкозы в почечных клетках.
Хотя известно, что увеличение синтеза ГК может быть особенностью диабетического гломерулосклерозиса, до настоящего времени известно относительно немного изменений в производстве ГК в почечных полостях. Интересно размышлять о функциональном значении увеличения производства ГК. Недавние исследования предполагают, что высокая молекулярная масса и низкая молекулярная масса олигосахаридов ГК предоставляют различные сигналы клеткам. В общем, производство ГК высокой молекулярной массы предполагает нормальное гомеостатическое состояние среды, тогда как производство фрагментов ГК низкой молекулярной массы сигнализирует разрушение нормальной гомеостатической окружающей среды. Несколько исследователей сообщили, что олигосахариды ГК могут стимулировать генную экспрессию и синтез белков хемокинов и промежуточных коллагенов. В контрастнее, ГК олигосахариды высокой молекулярной массы ингибируют "биологическую активность" TGF-? и стимулируют секрецию тканевых ингибиторов металлопротеиназ. Поэтому эти наблюдения предполагают, что ГК высокой молекулярной массы может быть "антифиброзным", тогда как постоянное производство ГК низкой молекулярной массы фрагментирует, может разрушить нормальный баланс между клетками и матриксом и поспособствовать патофизиологии хронического воспаления ткани и фиброза. Поэтому анализ молекулярной массы произведенного ГК может быть ключом к пониманию значения увеличения синтеза ГК, что мы описали.
Приток макрофагов был ранее вовлечен в патогенез диабетической нефропaтии, и в моделях животных, и в человеческих болезнях. Это предполагает, что производство полученных цитокинов макрофага, таких как IL-1?, в комбинации с эффектом повышенных концентраций глюкозы может действовать синергистически, чтобы влиять на патогенез диабетической нефропaтии. Мы ранее продемонстрировали, что объединенные эффекты глюкозы и IL-1? могут смодулировать профиброзный потенциал PTC в диабетической нефропaтии увеличением производства профиброзного цитокина TGF-?1. Позже увеличение активации NF-каппа В провоспалительными цитокинами при повышенных концентрациях глюкозы была продемонстрирована в сосудистых гладких мышечных клетках. В текущей рукописи мы продемонстрировали, что стимуляция с комбинацией IL-1? и 25 ммоль D-глюкозы имело совокупный эффект на производство ГК, как в дальнейшем получило поддержку мнение, что взаимодействие гипергликемии и макрофага приводит к тому, что провоспалительные цитокины могут увеличивать почечные раны при диабете.
В резюме мы продемонстрировали впервые, что увеличение синтеза ГК или в ответ на IL-1?, или в ответ на 25 ммоль D-глюкозы, зависит от NF-каппы В, которая активизирует транскрипцию ГКС2. Это может иметь большое значение для патогенеза почечных промежуточных изменений, связанных с диабетом меллита. Мы также продемонстрировали, что ГКС3 обязательно экспрессируется и не производит стимуляции в этих клетках, хотя роль обязательного ГК синтеза в этих клетках еще необходимо выяснить. Поэтому идентификация и противопоставление сигнальных путей, которые добиваются транскрипционной активации ГКС2 после стимуляции IL-1? или 25ммоль D-глюкозой представляют важную область для будущего исследования.
Подтверждения
SGJ поддержан Национальным Почечным Фондом Исследования, и AOP получил доверие Передового Учебного Товарищества.